

医疗器械注册证申报疝修补补片产品技术要求:
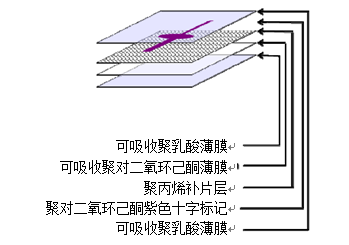
医疗器械产品注册证申请人/生产企业应依据具体产品的特性确定以下技术要求是否适用,若不适用需详细说明理由并提供支持性资料。
(1)外观及尺寸:长度、宽度、厚度、孔尺寸、适用的孔隙项目要求(如网孔密度/网孔比例/孔隙率)、特殊形状或结构所涉及的其他尺寸,包括允差。
(2)物理性能。
① 单位面积重量;
② 拉伸强度;
③ 顶破强度;
④ 缝合强度;
⑤ 若是多层结构或由不同部件连接的产品,要求制定连接强度;
⑥ 拉伸伸长率;
⑦ 撕裂强度。
(3)化学性能。
① 对于人工合成的不可吸收材料(如聚丙烯、聚四氟乙烯、聚酯等)制成的产品,应包括红外鉴别、酸碱度、还原物质、蒸发残渣、紫外吸光度、重金属总量、微量元素、终产品中有害小分子物质的残留量要求等。
② 医疗器械注册证代理对于人工合成的可吸收材料(如聚乳酸等)制成的产品,应包括红外或核磁鉴别、特性粘度或平均分子量、分子量分布(如适用)、旋光度(如适用)、单体残留、催化剂残留、溶剂残留、水分残留、重金属含量、终产品中其他有害小分子物质的残留量要求等。若材料为共聚物,还应要求共聚物中各单体形成结构单元的摩尔分数。
③ 医疗器械产品注册证代理对于由天然材料提取制备而成的可吸收材料,如胶原、纤维素等制成的产品,至少应包括材料定性要求、材料纯度要求、环境污染可能造成的重金属残留、终产品中有害小分子及大分子物质的残留量要求等。
④ 对于由动物或人体组织材料经处理制成的产品,如以真皮、小肠粘膜、肌腱、心包膜等组织为原料的产品,至少应包括环境污染可能造成的重金属残留、终产品中有害小分子及大分子物质的残留量要求等。
⑤ 对于经环氧乙烷(EO)灭菌的产品,应制定EO残留量要求。
⑥ 对于染色的补片,应制定褪色试验要求。
医疗器械注册证申报除上述要求外,还需参照相关材料的国家标准/行业标准增加适用的化学性能要求;化学性能试验浸提介质和浸提条件的选择应有充分的依据)
(4)无菌。
(5)细菌内毒素。
(6)若产品中含有致热性的材料成分,则需在产品标准中增加热原检测项目。
(7)对含有可降解/可吸收成分的产品,制定降解性能要求。
(8)对于动物源性材料制成的产品,应参照《动物源性医疗器械产品注册证申报资料指导原则》制定适用的技术要求。如免疫原性控制要求,可通过生物化学方法直接测定免疫原性指标,也可通过物理或化学方法测定某些指标来间接反映产品免疫原性。注册产品标准的编制说明中应给出制定这些具体指标及检测方法的科学依据,以证明产品的免疫原性被控制在可接受范围内。同种异体材料制成的产品也可参照上述要求。
(9)生物学评价。
医疗器械注册证代办应按照GB/T 16886《医疗器械生物学评价》进行生物学评价或试验,涉及项目如下(以现行有效的GB/T 16886.1为准):
① 细胞毒性;
② 迟发型超敏反应;
③ 遗传毒性;
④ 植入反应;
⑤ 全身急性毒性;
⑥ 刺激或皮内反应;
⑦ 亚慢性毒性;等。
(10)医疗器械产品注册证代办对于除补片外还包含其他部件及工具的产品,申请人/生产企业应制定相应部件或工具的项目要求。
鸿远医疗器械咨询 http://www.yixiezixun.com是一家技术专业的医疗器械产品注册证代办理咨询,医疗器械生产许可证代办,医疗器械经营许可证代办,一类医疗器械产品备案代办,二类医疗器械经营备案,CE认证,ISO13485认证,FDA注册,FDA认证,临床试验,医疗器械质量管理体系认证及体系建立与过程确认文件建立(ISO9001, ISO13485, GMP, CE,QSR820,CMDCAS);注册,出口证,自由销售证办理,临床试验及免临床资料编写,产品技术要求制订,技术文件编写辅导等提供一站式服务机构,欢迎您咨询与合作!
 在线客服
在线客服