

�� ҽ����е��Ʒע��ҽ�÷����(���¼�Ʒ����)����GB/T 4240�й涨��30Cr13��40Cr13��12Cr18Ni9��06Cr19Ni10�ƺŲ���ֲ�������,���ڷ�����ࡢ����֯��Ƥ���ȣ�Ϊһ����ʹ��
����ҽ�÷�����Ʒע��ע���걨����Ҫ��
����(һ)�����Ϣ
����1.�����
����1.1��Ʒ����Ӧ���ϡ�ҽ����еͨ���������������ұ�����ҵ���е�ͨ������Ҫ���磺����롢ҽ�÷����ȡ�
����1.2���ݡ�ҽ����е����Ŀ¼�����걨��Ʒ�������Ϊ02-07-01��
����1.3ע�ᵥԪ�Ļ���Ӧ�Բ�Ʒ�ļ���ԭ�����ṹ��ɡ�����ָ������÷�ΧΪ�������ݡ�
�����磺��ͬ�ƺŲ���ֲ��������ķ����ɻ���ͬһע�ᵥԪ��
����2.��Ʒ�б�
�����Ա�����ʽ�г��걨��Ʒ���ͺš���ṹ����ɡ��������Լ�ÿ���ͺŹ��ı�ʶ(���ͺŻ��ı�ţ���еΨһ��ʶ��)������˵��(��ߴ硢���ʵ�)��
����3.Ӧ���ա�ҽ����еע���걨����Ҫ��˵������Ҫ���ύ������������д���б��������ļ����걨ǰ���ܻ�������ϵ�����ͨ��¼(������)�����ĵ���Ȩ��(������)�������������ȡ�
����(��)��������
����1.����
�������������⡢���塢��β���(��ͼ1)���ò�Ʒһ��һ����ʹ�á�
�����������ʽ�����Ρ���ײ�ͬ��Ϊ������ʽ��ÿ����ʽ��ֱ�����ҳ����볤�IJ�ͬ��Ϊ��ͬ���
����������ʽ���֣�Բ�롢������(�����ǡ������ǡ��������ǡ����䡢���ǰ����)�����롢���롢��ʯ��ȡ�������ʽ��
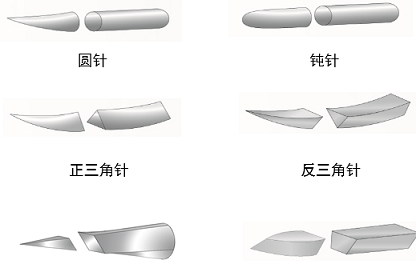
������������ ��������
������ʯ�� ����
�������ݻ��λ��֣�ֱ�Ρ�1/4����3/8����1/2����5/8���ȡ����ֻ���ʾ����ͼ3��
������������֣���ͨ�ס������롢�����ס����ʾ����ͼ4��
������������ʽ�����Ρ������������з�����ʾ��
��������
����1)��ʽ�������ֺ�/����ű�ʾ���£�
����Բ��“��”;
���������ǻ�“��”;
���������ǻ�“��”;
�����������ǻ�“���С�”;
��������;
�������ǰ����“������”;
�������λ�“ ”;
��������;
������ʯ��
����2)���Σ���ʾ���£�
����ֱ�Ρ�1/4����1/2����3/8����5/8����
����3)��ף��Դ��ű�ʾ���£�
����A(��ʡ��)——��ʾ��ͨ�ס�������;
����B ——��ʾ�����ס�
����4)���ʾ���£�
����ֱ��×�ҳ�����ֱ��×�ҳ�—�볤��
����(ֱ���Ա�ʾֵ×10��ʾ)
����ʾ����
����ֱ��D=0.7mm���ҳ�L=24mm���볤=31mm�����Ϊ��ͨ�ף���ʽΪԲ�룬����Ϊ3/8������ʾΪ��
����Բ3/8��·7×24����Բ3/8��·7×24—31;
������3/8��·7×24����3/8��·7×24—31��
����2.����ԭ��
����ͨ�����������ṹ���д��̡��и���������ƽ�����ɣ���������߽��з�ϡ�
����3.��װ����
����˵�����в�Ʒ��ɵİ�װ��Ϣ��Ӧ��˵����������ϵͳ����Ϣ��˵�����ȷ������ʹ���߿������ر�ʶ��װ�������ԡ�
����4.�����
�������������Ƥ�и��Ǻ������з�������Ŀ�ġ����вο���ͬ���Ʒ��ǰ����Ʒ��Ӧ���ṩͬ���Ʒ��ǰ����Ʒ����Ϣ����˵��ѡ������Ϊ�з��ο���ԭ��
����5.���÷�Χ
�������ڷ�����ࡢ����֯��Ƥ���ȡ�
����6.Ԥ��ʹ�û���
����Ԥ��ʹ�û���Ϊҽ�ƻ�����
����7.����֤
������δ���֡�
����8.�����¼����
������Ʒ��ʹ�ù��������ж���ȿ��ɲ����¼�������
����(��)���ٴ�����
����1.��Ʒ���չ�������
����ҽ����еע�����������ύ���չ������ϣ�ʶ����ҽ����е�йص�Σ��(Դ)�����ƺ�������ط��գ�������Щ���ղ����ӷ��տ��Ƶ���Ч�ԣ�����YY/T 0316��ҽ����е ���չ�����ҽ����е��Ӧ�á���Ҫ�����Ҫ�������
����(1)����ע���Ʒ�����ƽΣ��Ѷ����йؿ��ܴ��ڵ�Σ���������ķ��ս��й��ƺ����ۣ���������Ե�ʵʩ�˽��ͷ��յļ�����������Ĵ�ʩ��
����(2)�Ƿ���ȷʶ�����Ӱ��ҽ����е��ȫ�Ķ��ԺͶ����������γ��ļ�(�ο�YY/T 0316��¼C)��
����(3)Σ��(Դ)�����Ƿ�ȫ�棬������Ӧ��д������ʹ�úͷ�����ʹ�����������£����Ʒ�йص���֪�ĺͿ�Ԥ����Σ��(Դ)�ļ�(�ο�YY/T 0316��¼E)��
����(4)���տɽ������ͷ��յĴ�ʩ����ȡ��ʩ����յĿɽ��ճ̶ȣ��Ƿ����µķ��ղ�����
������2�о��˷�����Ʒ�����漰��Σ��(Դ)(����YY/T 0316��¼E)����ҵ��Ӧ����������Ʒ�ص�ȷ������Σ��(Դ)����Բ�Ʒ�ĸ�����գ���ҵӦ��ȡ���ƴ�ʩ��ȷ�����ս��͵��ɽ��ܵij̶ȡ�
| Σ�գ�Դ�� | ��Ԥ�����¼����� | Σ����� | �˺� |
|
����Σ�գ�Դ�� |
�������ʹ�ù������������Բ�����г�λ�ò���ȷ��ԭ����ɶ��� | ���� | �������ڻ������ڣ��������ƣ���в����������ȫ |
| �̴������и������� | ʹ�ù����в��״��̼���������������� | �������ƻ�����ʧ�� | |
| Ӳ�Ȳ��� | ���� | �������ƻ��Ʒ��ʹ�� | |
| ��Ʒ���ظ�ʹ�� | ��Ʒ���������½� | �������ƻ�����ʧ�� | |
| ��ѧΣ�գ�Դ�� | ԭ���ϲ��ϸ���ʴ������� | ���ϸ���Ϻͻ���ֱ�ӽӴ� | �����̼�������֢״��������ڻ������ڣ���в����������ȫ |
| �������ӹ�����ʹ�õ����и��������������Ӽ��������Ŀ���δ�ܰ���ȷ�ϣ���δ�ܰ�ȷ�ϵĽ��ʵʩ���ƣ���ʹ���Ӽ��������� | ���������Ӽ��������� | �������ϣ�Σ�����߿�����ȫ���������� | |
| �Ի�������������Ŀ���δ�ܰ���ȷ�ϣ���δ�ܰ�ȷ�ϵĽ��ʵʩ���ƣ����������ף���ʹ����������������� | �����Ļ�������������� | �������ϣ�Σ�����߿�����ȫ���������� | |
| δ�ܰ����䡢����Ҫ��Բ�Ʒʵʩ��������ɲ�Ʒ��װ���𣬲�Ʒ����Ⱦ | ����ʹ�ò��ϸ��Ʒ | ��ɸ�Ⱦ��/���Ⱦ�����»��ߴ̼���������֢���ݿˡ�����ʱ���� | |
| ����ѧΣ�գ�Դ�� | ʹ�����ﲻ���ݵIJ��� | ���ﲻ���ݲ����뻼��ֱ�ӽӴ� | ���»��ߴ̼���������֢���ݿˡ�����ʱ���� |
| �������δ�õ�ȷ�ϻ�δ����ȷ���Ĺ���ʵʩ��� | ���ϸ��Ʒ�뻼��ֱ�ӽӴ� | ���»��ߴ̼���������֢���ݿˡ�����ʱ���� | |
| ���������Ч�ڵIJ�Ʒ���� | ���ϸ��Ʒ�뻼��ֱ�ӽӴ� | ���»��ߴ̼���������֢���ݿˡ�����ʱ���� | |
| �ڲ�Ʒ�涨����Ч��ǰ����װ�����ϻ����ܱ�����ˮƽ | ���ϸ��Ʒ�뻼��ֱ�ӽӴ� | ���»��ߴ̼���������֢���ݿˡ�����ʱ���� | |
| ��Ʒ���ظ�ʹ�� | ����ʹ���о���Ʒ | ���»��ߴ̼���������֢���ݿˡ�����ʱ���� | |
| ��Ʒʹ����δ��ҽ���������� | �о����ж�����Ӱ�컷�� | ��ɻ���ҽ����Ա֮��ĸ�Ⱦ��/���Ⱦ����в����ҽ����Ա������ȫ | |
| ����Σ�գ�Դ�� | ���߱���Ʒʹ�����ʵ���Աʹ�ò�Ʒ���������� | ����ʹ�ò�Ʒ | ���²�Ʒ���ѡ�ϸ����Ⱦ���������ơ���в����������ȫ |
| ��ϢΣ�գ�Դ�� | �ⲿ���ڲ���Dz�ȫ�桢��Dz���ȷ���ܹ�������ϣ���ʶλ�ò�ǡ�����Լ���ʶ���ܹ��������� | ������Ա������ȷʹ�ò�Ʒ | ��ƷʧЧ����Ա���ˣ��ͺŹ��ѡ�ô�����ʧ�ܡ���ɴ̼���������֢���ݿˡ�����ʱ���� |
| ˵����ȱ�ٱ�Ҫ�ľ���˵������ϸ��ʹ�÷�����ȱ����ϸ���ճ�ʹ��ά���淶��˵�������й�ά�������������ݲ���ȷ | ������Ա������ȷʹ�ò�Ʒ | �������ƻ�����ʧ�� |
���������Ļ�����������ָ����Ҫ����YY/T 0043��ҽ�÷���롷���ڴ˻����ϣ�������Ӧ���ݲ�Ʒ���ص��ƶ���֤��Ʒ��ȫ��Ч�������ɿصļ���Ҫ�����ò���Ӧ���о�������˵�����ɡ���Ʒ����ָ�꼰���鷽����Ӧ������֤�������ļ���ָ��������������������ݣ�
����2.1���
����2.1.1����
����2.1.2�����п�
����2.1.3����ֲڶ�
����2.1.4������Ƕ��(������)
����2.2��������
����2.2.1Ӳ��
����2.2.2����
����2.2.3����
����2.2.4���ǿ��(�ҳ�L≥12mm�ķ��������)
����2.3ʹ������
����2.3.1�̴���
����2.3.2���
����2.4��ʼ��Ⱦ��(������)
����2.5��ʴ����
����2.6���(������)
����2.7�����������(������)
����3.ͬһע�ᵥԪ�ڲ�Ʒ���������ȷ��ԭ��Ͳ�Ʒ���鱨��
���������Բ�Ʒ��ָ�ܹ����DZ�ע�ᵥԪ��ȫ����Ʒ���յ�һ��������Ʒ��������Բ�ƷӦ���Dz�ͬ�ƺŲ���ֵIJ�Ʒ��
��������“ͬһҽ����е��Ʒע����Ԫ�ڣ������IJ�ƷӦ�����ܹ�������ע�ᵥԪ��������Ʒ��ȫ�Ժ���Ч�Եĵ��Ͳ�Ʒ”��ԭ��ȡ��ƷӦ�ܺ��Ǹ�ע�ᵥԪȫ����Ʒ�ļ���Ҫ����ͬһע�ᵥԪ���в�ͬ���͡���ͬ�ƺŲ���ֵķ���룬��Ӧ�ֱ���м��顣
����4.�����
�����������걨�IJ�Ʒ���ṩ��Ӧ���о����ϡ�
����4.1�����ͻ�ѧ�����о�
����ҽ����еע��֤������Ӧ���ṩ��Ʒ�����о������Լ���Ʒ����Ҫ����о��ͱ���˵�������������ԡ���ȫ��ָ���Լ�������������ص�����ָ���ȷ�����ݣ������õı���������ԭ�����ۻ�����
��������YY/T 0043��δ���ǵ����ͣ�Ӧ��ϲ�Ʒ�ص��Ԥ�ڷ�ϲ�λ�������ƶ������õ�����ָ�꣬��Ӳ�ȡ��̴������и�����ָ�ꡣ
������Ʒ�ļ��鷽��Ӧ���ݼ�������ָ���趨�����鷽��Ӧ���Ȳ��ù��ϵĻ��Ѱ䲼�ı����鷽�������û�����еı����鷽���ɲ���ʱ���涨�ļ��鷽��Ӧ���пɲ����ԺͿ������ԣ���Ҫʱ��ȷ��Ʒ���Ʊ���������Ҫʱ�ɸ���Ӧͼʾ����˵�����ı��ϴ�Ŀ��Ը�¼��ʽ�ṩ��
����4.2���������������о�
����ҽ����е��Ʒע��֤����Ӧ����GB/T 16886.1��ҽ����е����ѧ���� ��1���֣����չ��������е����������顷�Գ�Ʒ(��Ҫ�����뻼�ߺ�ʹ����ֱ�ӻ��ӽӴ��IJ���)�����������Խ������ۡ����������������о�����Ӧ���������������������۵����ݺͷ�������Ʒ���ò��ϵ�������������Ӵ������ʣ�ʵʩ���������ѧ��������ɺ���֤�������������ݻ������������ۡ�
��������ѧ���۹�����Ӧ��ע������������Ϣ(�������ϡ��������ϡ�����������������ݡ��ٴ�����)��������ѧ����ȷ����Ҫ��������ѧ����ʱ��Ӧ���ٷ���YY/T 0043����Ҫ��ϸ���������鷴Ӧ������1��;�ٷ��ͳ�����ӦӦ������1��;Ƥ�ڷ�Ӧ�Ʒ�Ӧ������1��
����4.3��������о�
����4.3.1������ҵ�����Ӧ��ȷ�������(�����Ͳ���)������֤ˮƽ(SAL)�����ṩ���ȷ�ϱ��档��е�����Ӧͨ��GB 18279.1��GB/T 18279.2��GB 18280.1��GB 18280.2��GB/T 18280.3ȷ�ϲ����г�����ƣ�����֤ˮƽ(SAL)Ӧ��֤�ﵽ1×10-6��
����4.3.2�ն��û������Ӧ����ȷ�Ƽ����������(�����Ͳ���)�����Ƽ����������ȷ�������ݡ��ն��û�����ķ��������֤ˮƽ(SAL)ҲӦ�ﵽ1×10-6��
����4.3.3�������ԣ������ʹ�õķ������׳��ֲ���������û������������Ӧ����ȷ��������Ϣ����ȡ�Ĵ������������ṩ�о����ϣ���ҵ���ṩ��֤��Ʒ����ʱ����������������ô���10μg/g�Ĵ���������
����4.3.4������ƣ�ͨ�����������������������̱�֤��ʼ��Ⱦ��ˮƽ��������������ṩ�����ʼ��Ⱦ��ӦС�ڵ���100 CFU/��;��Ӧ���ṩ֤����װ�ܼ��ٲ�Ʒ�ܵ�������Ⱦ�ķ��գ���������������ҵ�涨����������о����ϡ�
����4.3.5������̵�ѡ��Ӧ���ٿ����������أ���Ʒ��������̼����Ӧ��;��װ������������̵���Ӧ�ԡ�����Բ�Ʒ��ȫ��Ч�Ե�Ӱ��ȡ�
����4.4��Ʒ������Ч�ںͰ�װ�о�
����4.4.1������Ч�ڰ�����Ʒ��Ч�ںͰ�װ��Ч�ڡ���Ʒ��Ч����֤�ɲ���ʵʱ�ϻ�������ϻ����о��������ϻ������о��ľ���Ҫ��ɲο�YY/T 0681.1��Ӧ��ѭ���������ԭ���ڽ��м����ϻ������о�ʱӦע�⣺��Ʒѡ��Ļ����������ϻ�����Ӧ�����Ƶ����䴢����ʵ�����·�����Ʒ�ϻ��Ļ�����ƥ��һ�¡�
�������ڰ�װ����Ч����֤�������������ύ��ѡ��ǡ���IJ��ϺͰ�װ�ṹ�ϸ������ճ�Ʒ��װ�ij�ʼ�����Ժ�ά�������Եļ������
����4.4.2��װ����װ�����ԣ������Ƶ���Ч�����Լ����䴢�������£����ְ�װ�����Ե����ݡ���װ���ϵ�ѡ��Ӧ���ٿ����������أ���װ���ϵ�������ѧ����;��װ���ϵĶ���ѧ����;��װ�������Ʒ����Ӧ��;��װ��������ͺ��ܷ���̵���Ӧ��;��װ������������̵���Ӧ��;��װ���������ṩ����������ѧ���������ϱ���;��װ������ʹ����ʹ��ʱ��Ҫ��(��������)����Ӧ��;��װ�������ǩϵͳ����Ӧ��;��װ������������������;�����̵��ʺ��ԡ��ɲο�GB/T 19633��YY/T 0681ϵ�б��ȡ�
����5.��������
����֤����Ʒ��ȫ�ԡ���Ч�Ե������о����ϡ�
����(��)�ٴ���������
�����ò�Ʒ���롶�����ٴ�����ҽ����еĿ¼���������������ύ�ٴ��������ϡ�
���������롶�����ٴ�����ҽ����еĿ¼�������������ҽ�÷�����Ʒ��Ӧ���ա�ҽ����е�ٴ����ۼ���ָ��ԭ��Ҫ���ύ�ٴ��������ϡ�
����(��)��Ʒ˵����ͱ�ǩ����
������Ʒ˵����ͱ�ǩ�ı�дӦ���ϡ�ҽ����е˵����ͱ�ǩ�����涨����Ҫ���ɲο�YY/T 0043��ҽ�÷���롷��YY/T 0171�������е ��װ����־��ʹ��˵���顷��YY/T 0466.1��ҽ����е ����ҽ����е��ǩ����Ǻ��ṩ��Ϣ�ķ��� ��1���֣�ͨ��Ҫ����ر������ύ���ı��ͱ�ǩ����Ӧ����������������˵�����е����÷�Χ������֤��ע�������ʾ��Ϣ����Ч�ڵ���ϢӦ���Ʒ�������ϡ��о����Ϻ��ٴ���������������������֤������һ�¡�
����ҽ����еע�������Ʒ˵������Ӧע����Ʒ�Ľ���״̬;Ӧע��ʹ��ʱһ�����������ǯ�����ʱӦ��ע�����ʹ�õ�Ҫ��;Ӧע����ƷΪ���������ʱӦ��ע���װ���ϡ���װ��ʽ��ʹ�÷�ʽ˵����ע��������ռ������ﴦ�á���Ʒ�ͺŹ��϶࣬Ӧ��ע���ǩ�Ƿ��ܹ�ָ��ʹ����Ա��ȷѡ�á�
����ͬʱ����˵�����ע�������ʾ��Ϣ��Ӧ���ٰ����������ݣ�
����1.��ƷӦ�������ʵ�ҽ����Աʹ��;
����2.��Ʒ��ʹ�������ڼг�λ�ò���ȷ��ԭ����ܵ��¶���;
����3.���ֻ��߶Է������Ͽ��ܲ�������֢״;
����4.��Ʒʹ�ú�Ӧ����ҽ�ƻ��������������淶���д���;
����5.���ڵ����ײ�Ʒ����Ƕ��ʱӦ����������ߡ����ߵ����⡣
����(��)ҽ����еע��֤��������������ϵ�ļ�
����1.�����˻�������������걨��Ʒ�ж�����ơ��������أ�Ӧ����ÿ�����ơ��������ص�ʵ�������
����2.��������֯����ͼ��
����3.������ҵ��ƽ�沼��ͼ����������ֲ�ͼ��
����4.���������о���Ҫ��Ӧ���ṩ�����ʵļ��������ߵĻ�����ⱨ��(��ƽ�沼��ͼ)��ӡ����
����5.�����ԭ����Ӧ����YY/T 0043��ҽ�÷���롷Ҫ��Ӧ������Ʒ���������������ԭ�������ơ���Ӧ�����ơ����ϵı��Ȼ�����Ϣ����Ҫԭ����Ӧ�����ȶ��Ĺ��������Ա�֤��Ʒ���������ṩԭ�����������ҵ�����֤�����Э�顣Ӧ��ȷ����ԭ����(��������װԭ����)����������Ҫ��
������ʹ���������ϣ�Ӧ�����䰲ȫ��Ч�ԡ�
����6.ҽ����е��Ʒע�����Ӧ�ύ��Ʒ���������չ��������ļ�����ϸ˵����Ʒ���������պͲ��裬�г�����ͼ���������������չ��̵��������Ʊ������ƴ�ʩ��һ�������������Ҫ������ӹ�(������˿��ֱ�����ϡ�ĥ�⡢�⡢ѹ�⡢���/��ס�ѹ�ǡ�ɰ�ǡ������)���������(�����ȴ��������洦����)�ͳ�Ʒ����(����ĩ����ϴ����ɡ�����軯����װ��ڡ������)��
����Ӧע��������̺ؼ�����(���ȴ��������洦����ĩ����ϴ����װ��ں������)����ȷ����̿��Ƶ㼰���Ʋ��������������յĿɿ��ԡ��ȶ���Ӧ����ȷ�ϡ�
�����������ӹ�����ʹ�õ����и��������������Ӽ�(���磬�����������������軯ʱ��������ʹ�õĹ��ͺ���ϡ�ͼ�)��Ӧ˵��ʹ�ü������Բ������Ŀ��ƴ�ʩ�ͽ��ܱ��Լ���ȫ�����۱��档
����7.��Ҫ�����豸�ͼ����豸(�����������顢���̼��顢�������ռ������������豸����������豸)Ŀ¼��
����8.����������ϵ�Բ鱨�档
����9.Ӧ���ṩ��˲��Ʒ�������ͨ���˲��Ʒ�������������������յȷ���ĶԱ�˵��(������)��
��Զҽ����е��ѯ����˾ ��һ�Ҽ���רҵ��ҽ����е��ѯ����˾��רע�ṩȫ�������磺���ڡ����ݡ���ݸ����ɽ����ɽ�����ݡ�˳�¡��������Ϻ������������졢�ɶ������ա����ա��㽭��֪�����е�ҽ����е��������ѯ����Զҽ����е��ѯרҵ�����ڣ�ҽ����е��Ʒע��֤��������ѯ��ҽ����е��Ʒ����綨������,����ҽ����е��������֤��һ��ҽ����е��Ʒ�������졢��������Լ�ע�ᡢ����ҽ����еע�ᡢҽ����е��Ӫ����֤���졢����ҽ����е��Ӫ������CE��֤��ISO13485��֤��FDAע�ᡢFDA��֤���ٴ����顢ҽ����е����������ϵ��֤����ϵ���������ȷ���ļ�����( ISO13485, GMP, CE��QSR820��CMDCAS);ע�ᡢҽ����е��������֤������Ʒ����Ҫ���ƶ��������ļ���ͬ���Ʒ�Ա����ٴ��������ϱ�д��ע�����ϱ�д������ҽ����е��������걨����ż������ĵ��ṩһվʽ����˾����ʦ�ż��ֻ��ţ�13590396780��ӭ����ѯ�������
 ���߿ͷ�
���߿ͷ�